成功开发合成脂类药物:关键方面和策略

由于在RNA治疗药物和疫苗开发领域,癌症和Covid-19等疾病中的重要作用,脂质正在增强势头。
目前,有18种脂质体药物,由美国食品和药物管理局(FDA)和临床中的数百种基于脂质的药物候选者进行一系列疾病1.脂质的制剂和脂质纳米颗粒在药物开发和交付中表明了许可,因为它们的能力2,3:
- 通过保护API免受免疫反应、蛋白酶和其他因素的影响,提高活性药物成分(API)的稳定性
- 提高水溶性较差药物的溶解性和生物利用度
- 被动地瞄准因血管渗漏而发炎或肿瘤组织
- 改善陷阱API的毒性剖面
- 能够递送复杂的api,如RNA,容易不稳定,核酸酶介导的裂解,强烈的免疫反应,并不能到达作用位点。
查看视频中的简短概述:
The newest advancements in lipid-based drug delivery research and drug development is in the field of nucleic acid delivery, for APIs such as short RNAs for gene silencing or activation (for e.g. siRNA, miRNA, saRNA) and long RNA (mRNA) for applications in cancer therapy, enzyme replacement therapy, vaccines, and more. The first vaccine to enter clinical trials forcovid-19是mRNA疫苗在这里,病毒抗原的mRNA被包裹在脂质纳米颗粒中。新型冠状病毒RNA疫苗的成功将促进这一领域的发展,未来几年将批准更多使用脂质的基因治疗药物。
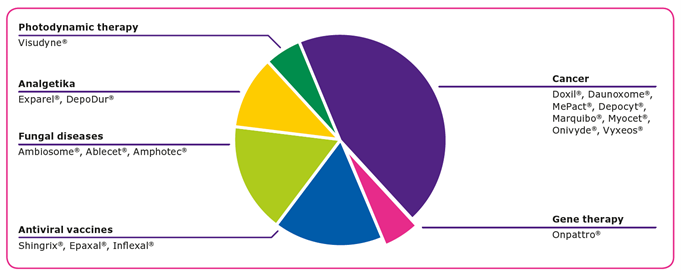
合成脂类的优点
脂质型,来源和质量/纯度对最终脂质体配方的杂质型材和性质进行直接影响,例如颗粒特征,双层结构,稳定性和药物释放曲线。对于可重复的结果,对于仅使用具有最佳材料特性和一致的质量,仅使用高质量的原料合成脂质。
化学合成的脂质在天然脂质上是有利的,因为它们由单一的已知质量脂质组成,而组织衍生的脂质通常通常是蛋衍生或牛衍生的脂质的混合物。与组织衍生的脂质不同,合成脂质不显示批量批量变异性或病毒或蛋白质污染的风险。
通过选择高质量的起始材料并优化制造工艺和纯化技术,可以优化合成脂质的纯度。原料应具有低水平的副产物,定义的立体化学(D / L)和异构纯度(CIS / Trans),低生物管和内毒素水平,并具有植物衍生的牛海绵状脑病(BSE)/传染性海绵状脑病(TSE)和非转基因生物(GMO)证书,并仅使用II类或III溶剂制备。应根据国际委员会统一的技术要求(ICH)在ICH Q3C中统一的技术要求的指导方针,避免载重贷款。
材料特性和可扩展性
GMP制造脂质体的药品制造需要高且一致的质量原料,以及包括溶解度,结晶度,稳定性和流动性的特性在制造过程中起重要作用。脂质,用于产生脂质体的关键原料是自然蜡状,这可能导致缓慢的溶解率并导致在大量处理时挑战。
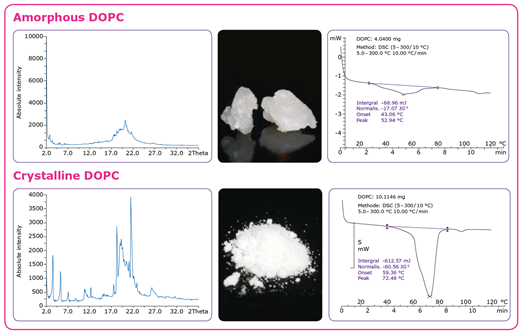
四种方法可用于增强脂质体的表面特性,以实现快速和完全溶解,并使脂质体制造过程可重复:冷磨,喷雾干燥,结晶和冻干。除了提高溶解度之外,这些加工方法还提供了更高的纯度、稳定性和更容易处理的特性,所有这些都使脂质体更容易配制。喷雾干燥和冻干的结果是物料具有非常高的表面积,高均匀性和良好的处理特性。结晶是最常用的方法之一,以提高表面脂质。
可扩展性、可重复性和GMP生产工艺的优化,包括脂质体的产量、浓度、同分异构体纯度和其他质量方面,以及反应和检测时间,对于确保最大的效率和最低的成本也是至关重要的。从一开始就应该考虑合成和纯化操作的可扩展性,以确保随着批规模的增加而实现规模经济,目标是减少合成步骤的数量,并为GMP生产明确定义它们。理想情况下,结晶或液/液萃取方法应用于纯化,只要可能,硅胶过滤可替代色谱。
脂质体制造方法
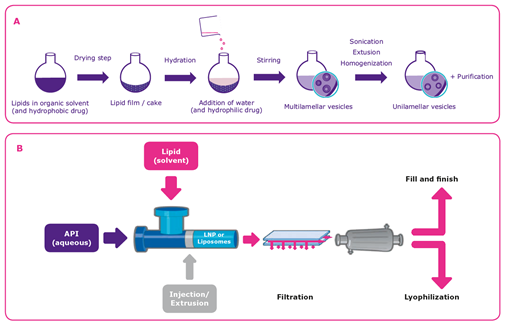
脂质体或脂质纳米颗粒可以用几种不同的方法制造,但挑战是确保可扩展的、健壮的和有效的过程4.一种制备多硅囊泡(MLV)(图3A)的方法是将脂质溶于有机溶剂中,然后在搅拌下用水去除(干燥)和水化。如果需要不硅烷囊泡(ULV),则可以通过添加声音或挤出步骤来减小囊泡的挤压步骤来产生它们。在溶解步骤期间将疏水API加入到溶剂中,而亲水性药物物质被添加到水合步骤中的水溶液中。纯化通常是该过程中的最终步骤。乙醇注射,其中脂质溶解在乙醇中并用含有API的水性介质快速混合,是制备含有亲水化合物的小ULV的突出替代方法(图3B)。
制造方法的选择通常取决于最终应用.6乙醇注射方法适用于生产小ULV和稳定的核酸脂颗粒,但不用于产生大型脂质体,例如施用较大的MLV和用于疫苗的较大的MLV和多重囊泡通过皮下注射或肌内注射。在这种情况下,使用再水解方法。
开发脂质的药品
在基于脂质体的药物发育的临床前阶段,应识别出最佳,经济高效的合成路线,完成可行性研究,并进行实验室规模的生产。过程优化应该是晚期临床前阶段和I期临床试验期间的重点,包括鉴定关键原料和脂质所需分析方法的开发。应在临床试验期间酌情进行适当的过程扩大,包括实施分析方法和过程内对照的测定,稳定性研究也在进行中。必须定义该过程的关键参数,在晚期临床试验期间,必须定义该过程的初步计划,原料供应商合格,严格的风险分析和中间体测试。选择错误的原材料和材料供应商可以导致负面的财务影响和延误。
基于脂质的药物制剂的监管方面
由于对脂质辅料缺乏全球统一的监管要求,脂质体药品的监管审批没有明确的途径。由于脂类成分的纯度和质量会影响脂基制剂的质量,监管当局要求提供有关化学、制造和控制的详细信息5-19.
由于具有挑战性的监管环境,建议药品制造商与供应商密切合作,在临床开发和商业化的所有阶段提供监管专业知识和咨询,涵盖质量保证和文件记录的所有方面。
结论
对于用合成脂质的成功的药物开发,GMP制造过程需要在质量和产量中可扩展和可重复。这是最终产品一致质量的先决条件。所用脂质的质量对脂质体配方的性能产生了重大影响。脂质以不同的形式,物理状态和来自不同供应商的纯版提供,并且根据应用,选择具有最佳特性的右脂质。在使用任何新型脂质的新脂质时,可行性研究以找到最佳的合成途径和纯化步骤,可以扩大到GMP生产是关键。
虽然脂质体药物产品的监管过程与来自不同全球当局的许多不同指南复杂,但它们都同意脂质质量至关重要。
为了避免药物开发过程中的高成本和意外,提前计划产品开发并在整个药物开发过程中使用相同质量的赋形剂是很重要的。这强调了与正确的供应商合作的重要性,提供一致的高质量产品,了解药物开发过程的所有步骤和监管环境,并提供高水平的客户支持。
Pharmaexcippers.com的说明性博客帖子 - 由Merck Kgaa,Darmstadt作者Shiksha Mantri,德国。默克公司(Merck KGaA)的生命科学业务位于德国达姆施塔特(Darmstadt),在美国和加拿大的业务名为MilliporeSigma。-版权所有
Shiksha Mantri是Merck Kgaa,德国达摩尔·克格纳的合成脂质的全球技术产品经理,负责管理整个GMP脂质业务(投资组合和定制制造)。她还支持各种默克倡议作为几个生命科学主题的主题专家。
要求更多信息或合成脂质样本:
参考
- Bulbake等。脂质体制剂在临床中的应用:最新综述。药剂。2017; 9(2)。网络。
- yingchoncharoen,Phatsapong,Danuta S. Kalinowski和Des R. Richardson。“癌症治疗中基于脂质的药物递送系统:有什么可用的,还有什么时候到来。”药理的评论。68 701-787(2016年)。
- Shrestha, Hina Rajni Bala和Sandeep Arora。“脂质药物传递系统。”制药学杂志》.2014年5月19日。网络。
- 瓦格纳和k·沃罗尔-乌尔。“工业用途的脂质体技术。”毒品混合。2011: 591325(2011)。
- 食品和药物管理局(FDA)脂质体药品:化学,制造和对照;人类药代动力学和生物利用度;和标签文档。2018年4月。网。
- 11-11药物物质(化学实体和生物技术/生物实体)的开发和制造。国际人用药品技术要求协调理事会(ICH)。2012年5月1日。网络。
- “关于静脉注射脂质体产品的数据要求参考创新脂质体产品的数据要求。”欧洲药物局(EMA)。2013年2月。网。
- 脂质体药品开发指南。厚生劳动省(MHLW)。2016年3月。网络。
- ICH Q7(API GMP):活性药物成分Q7的良好制造实践指南。国际的人用药品技术要求协调委员会(ICH)。10 2014年11月。网络。
- ICH Q1A(R2):新药物和产品的稳定性测试.国际人用药品技术要求协调理事会(ICH)。2003年6月。网络。
- ICH Q2(R1):分析方法的验证:文本和方法学.国际人用药品技术要求协调理事会(ICH)。2005年11月。网。
- Q6A规范:新药和新药的试验程序和验收标准:化学物质。国际人用药品技术要求协调理事会(ICH)。6 1999年10月。网络。
- 《药物辅料联合良好生产规范指南》国际药物辅料理事会(IPEC)和药物质量集团(PQG)。2017.网络。
- "药品辅料良好分配规范指南"国际药物辅料理事会(IPEC)。2017.Web . .
- 药品用辅料资格鉴定.国际药物辅料理事会(IPEC)。2008.网。
- “iPec辅料稳定性节目指南。”国际药物辅料理事会(IPEC)。2010.网。
- “iPec赋形剂组成指南。”国际药物辅料理事会(IPEC)。网络。
- “药物赋形剂的IPEC重大变化指南。”国际药物辅料理事会(IPEC)。2014.网。
- “Ipec-Americas expipient Master文件指南”。国际制药辅料委员会